La bioinformática es el área de la computación que tiene mayor potencial para transformar el estado de la Humanidad a corto-medio plazo. Es escasamente comentada por los medios de comunicación, probablemente debido a la inexistencia de productos de consumo masivo con fuertes inversiones en campañas de publicidad y relaciones públicas.
La bioinformática es el área de la computación que tiene mayor potencial para transformar el estado de la Humanidad a corto-medio plazo. Es escasamente comentada por los medios de comunicación, probablemente debido a la inexistencia de productos de consumo masivo con fuertes inversiones en campañas de publicidad y relaciones públicas.
Cuando se habla de bioinformática se piensa casi siempre en la secuenciación de ADN. Pero los desafíos científicos de la bioinformática no se limitan a la manipulación del código genético, sino que abarcan otras áreas como la predicción de estructura y alineamiento estructural de proteínas, el modelado de procesos evolutivos y, en general, la explicación de todos los procesos biológicos.
En este artículo voy a comentar sólo la genómica computacional, es decir, el estado del arte de la tecnología para tratamiento informatizado del código genético usando técnicas algorítmicas, estadísticas, y «Big Data». Pero el desafío científico es mucho más amplio e incluye:
• Cómo leer rápidamente, con exactitud y a bajo coste el ADN de un organismo vivo.
• Cómo almacenar las secuencias de ADN leídas.
• Cómo comparar nuevas secuencias con secuencias ya conocidas para establecer relaciones evolutivas entre ellas o encontrar mutaciones patógenas.
• Cómo explicar la síntesis de proteínas, crecimiento de tejidos, formación de órganos, coordinación entre órganosy otros procesos vitales con un lenguaje de alto nivel que nos permita describir organismos vivos.
• Cómo crear cadenas de ADN y cromosomas ensamblando genes que cumplan una determinada función.
• Cómo insertar el ADN generado en una célula capáz de producir la función especificada.
• Cómo alterar los genes ya presentes en un organismo vivo para corregir errores y curar enfermedades.
Es frecuente asociar la genómica computacional con el diagnóstico y tratamiento de trastornos de salud cuyo origen es genético, pero la genómica computacional es aplicable a muchas más cosas además de la medicina. Quienes están invirtiendo activamente en la recopilación y explotación de información genética tienen en mente un uso comercial mucho mayor. Actualmente aún es relativamente caro secuenciar ADN, pero llegará un día no muy lejano en el que las empresas paguen por tener nuestra información genética con el fin de usarla para personalizar su oferta, no sólo en forma de medicamentos a la medida del paciente, sino en base a cualquier cosa que el ADN pueda revelar acerca de las preferencias y necesidades de cada persona.
Como en ocasiones anteriores, he estructurado el texto desde lo menos técnico a lo más técnico. Necesito empezar explicando un poco el contexto de qué es el ADN y cómo funciona antes de poder comentar los métodos computacionales. Finalmente explicaré el hardware y otras tecnologías que se usan modernamente para la implementación.
Composición y estructura del ADN
Desde el punto de vista de el tratamiento de información, las cadenas de ADN son básicamente un lenguaje que explica cómo se sintetizan proteínas a partir de veinte aminoácidos constituyentes. El orden en el que se concatenan los aminoácidos determina la estructura de la proteína (que puede llegar a ser bastante compleja) y la estructura determina su función. Las proteínas sintetizadas forman células, las células forman tejidos, los tejidos forman órganos y los órganos cooperan para hacer funcionar el cuerpo humano.
 Las cadenas de ADN se forman con cuatro nucleótidos: dos purinas, adenina (A) y guanina (G) ; y dos pirimidinas, citosina (C) y tiamina (T). En adelante nos referiremos a estos nucleótidos simplemente como A G C T. Los nucleótidos se enlazan siempre en los mismos pares purina-pirimidina A-T y C-G dentro de la conocida estructura de doble hélice. Cada triplete de nucleótidos forma un codón. Un codón codifica un aminoácido. Hay 64 codones posibles, pero sólo 20 aminoácidos. Un aminoácido puede ser codificado por 1,2,3,4 o 6 codones distintos. También existe un codón de inicio y tres codones de terminación. Por ejemplo, la secuencia que da nombre a la película GATTACA podría interpretarse como GAT TAC A en cuyo caso codificaría ácido aspártico seguido de tirosina más una A extra; o como G ATT ACA y entonces sería una G seguida de isoleucina y treonina.
Las cadenas de ADN se forman con cuatro nucleótidos: dos purinas, adenina (A) y guanina (G) ; y dos pirimidinas, citosina (C) y tiamina (T). En adelante nos referiremos a estos nucleótidos simplemente como A G C T. Los nucleótidos se enlazan siempre en los mismos pares purina-pirimidina A-T y C-G dentro de la conocida estructura de doble hélice. Cada triplete de nucleótidos forma un codón. Un codón codifica un aminoácido. Hay 64 codones posibles, pero sólo 20 aminoácidos. Un aminoácido puede ser codificado por 1,2,3,4 o 6 codones distintos. También existe un codón de inicio y tres codones de terminación. Por ejemplo, la secuencia que da nombre a la película GATTACA podría interpretarse como GAT TAC A en cuyo caso codificaría ácido aspártico seguido de tirosina más una A extra; o como G ATT ACA y entonces sería una G seguida de isoleucina y treonina.
Animaciones de biología invisible, Drew Berry

Un grupo de genes capaces de ejercer una regulación de su propia expresión por medio de los sustratos con los que interactúan las proteínas codificadas forma una unidad genética denominada operón. Cada operón tiene tres partes: el factor promotor, el operador y el gen regulador en cuyos detalles no entraremos aquí.
Además del orden de la secuencia de nucleótidos, los hallazgos científicos recientes confirman que la forma en la que está plegado el ADN también es relevante para su funcionalidad. El genoma humano completo tiene unos 3.000 millones de pares de nucleótidos repartidos en 23 cromosomas. Puesto en línea mediría aproximadamente dos metros. Sin embargo, cabe dentro de los 10 micrometros del núcleo de cada célula porque está retorcido en nucleosomas. De la expresión de los genes que no depende de la secuencia sino de su plegamiento y otros factores se ocupa una rama llamada epigenética.
Hasta aquí lo que tenemos es una maquinaria de producción de proteínas y autoreplicación. Pero el ADN no puede ser sólo una superestructura molecular autoreplicante y generadora de proteínas. Tiene que ser algo más. El ADN debe poseer las cinco características de todo lenguaje: alfabeto, gramática, significado, intención y redundancia y corrección de errores.
Por poner sólo uno de los innumerables ejemplos que podríamos encontrar. Durante la reproducción los espermatozoides encuentran el óvulo debido a que éste emite calcio. Los espermatozoides pueden detectar el gradiente de calcio y nadar en la dirección en que aumenta la concentración. Este comportamiento tan complejo de una sola célula no puede explicarse sólo en términos de síntesis protéica. Tiene que existir algo más en el ADN que controle cómo nadan los espermatozoides.
Técnicas de secuenciación de ADN
Como síntesis del análisis anterior sobre la composición y estructura del ADN podemos decir que en los humanos encontramos tres mil millones de bits (nucleótidos) agrupados en palabras de 3 bits (codones) que componen subrutinas (genes) contenidas en módulos (operones). Pero por ahora nadie sabe cómo proporcionar una explicación de alto nivel a cómo interpretar este galimatías.
 El primer desafío es secuenciar el ADN. No se conoce ninguna técnica para leer un cromosoma de principio a fin. Lo que se hace actualmente se conoce como secuenciación por perdigonada seguida de un ensamblado de secuencias. Esta técnica consiste en cortar múltiples copias del ADN a secuenciar en múltiples fragmentos de una longuitud variable entre 600 y 800 nucleótidos. Luego averiguar cómo encjan unos fragmentos con otros casando el final de un fragmento con el principio de todos los otros maximizando el grado de solapamiento de nucleótidos.
El primer desafío es secuenciar el ADN. No se conoce ninguna técnica para leer un cromosoma de principio a fin. Lo que se hace actualmente se conoce como secuenciación por perdigonada seguida de un ensamblado de secuencias. Esta técnica consiste en cortar múltiples copias del ADN a secuenciar en múltiples fragmentos de una longuitud variable entre 600 y 800 nucleótidos. Luego averiguar cómo encjan unos fragmentos con otros casando el final de un fragmento con el principio de todos los otros maximizando el grado de solapamiento de nucleótidos.
| Secuencia original | AGCATGCTGCAGTCATGCTTAGGCTA |
| Primera perdigonada | AGCATGCTGCAGTCATGCT——- ——————-TAGGCTA |
| Segunda perdigonada | AGCATG——————– ——CTGCAGTCATGCTTAGGCTA |
| Secuencia reconstruida | AGCATGCTGCAGTCATGCTTAGGCTA |
Para complicar aún más el proceso, ningún secuenciador de ADN proporciona lecturas correctas con un 100% de probabilidad. Cada nucleótido es leído sólo con un porcentaje de probabilidad. Aunque con los secuenciadores más modernos y múltiples lecturas con consenso la probabilidad llega al 99,9%.
Similitud de secuencias de ADN
La variación genética entre humanos se estima que es entre el 0,1% y el 0,4% de los nucleótidos. Es decir, entre el ADN de dos humanos cualesquiera difiere, más o menos, en uno de cada mil nucleótidos. Cuando se secuenció el genoma humano por primera vez no se hizo con el ADN de un sólo individuo sino con el de cinco individuos. Estudios posteriores con 1.000 individuos de 26 poblaciones diferentes han mostrado que existen entre 4,1 y 5 millones de diferencias con el genoma de referencia. Debido a que, como ya hemos expuesto, dos codones diferentes pueden codificar el mismo aminoácido, muchas de estas variaciones son irrelevantes. Pero otras pueden causar graves enfermedades sólo por la variación de un nucleótido que imposibilita la síntesis de una proteína esencial para algún proceso vital.
Según las cifras que hemos indicado, los algoritmos de detección de disfunciones de origen genético deben buscar en unos tres millones de diferencias repartidos entre tres mil millones de pares.
Lo primero que se necesita pues es una métrica de similitud de secuencias. En genómica la similitud de secuencias de nucleótidos se mide comparando alineamientos. Para poder realizar un alineamiento de dos secuencias primero deben tener la misma longitud pues de otro modo no es posible hablar de los nucleótidos en la posición 1, 2 etc. Si dos secuencias no tienen la misma longuitud entonces se pueden insertar espacios al principio, al final o en medio de cada secuencia. Los espacios se conocen en la jerga genómica como “indels” (inner insertion or deletion) Por ejemplo, supongamos que empezamos con las secuencias GACTCT y GAGGC. Un posible alineamiento para ellas es:
GA_G_C_
Entonces una posible métrica es considerar el número de posiciones con el mismo nucleótido y restarle las posiciones con nucleótidos diferentes o con un espacio. En este caso las secuencias alineadas coinciden en los nucleótidos 1,2,6; difieren en la posición 3 y tienen espacios en 4,5,7. Por consiguiente, la puntuación para este alineamiento será 3-1-3 = -1. Tres coincidencias menos una no-coincidencia menos tres espacios. Con esta métrica, nuestra definición de lo que es el mejor alineamiento A posible de dos secuencias S₁ y S₂ será:
Por supuesto esta sencilla métrica no es la única válida. Lo que se deseará es emplear una métrica que devuelva la máxima similutud para secuencias de nucleótidos que tengan la misma función biológica aunque las secuencias no sean idénticas nucleótido por nucleótido. Es posible otorgar diferente peso a las no-coincidencias y a los espacios. También es común el uso de mutaciones puntuales aceptadas, es decir, substituciones en nucleótidos que se sabe que no afectan significativamente a la función de la proteína descrita por un gen. Pero por ahora no entraremos en las matrices de substitución y nos centraremos en los algoritmos de alineamiento de secuencias.
Alineamiento de secuencias de ADN
Los algoritmos más populares para alineamiento de secuencias son el Needleman-Wunsch para alineamiento global, el Smith–Waterman y el Altschul-Erickson para el alineamiento local y BLAST o FASTA para búsquedas heurísticas. El Needleman-Wunsch sirve para comparar si dos genes tienen probablemnente la misma función. Las variantes de Smith–Waterman se emplean para buscar subsecuencias y para averiguar si el final de una secuencia encaja con el principio de otra tal y como requiere el ensamblado de fragmentos en la secuenciación por perdigonada. BLAST y FASTA se usan para buscar rápidamente en una base de datos secuencias potencialmente similares a una dada y con los resultados preliminares devueltos por la búsqueda heurística de BLAST se ejecuta Needleman-Wunsch para encontrar la secuencia conocida más similar.
Veamos porqué se necesitan algoritmos eficientes para encontrar el mejor alineamiento de dos secuencias. Sea r la diferencia de longuitud entre las dos secuencias S₁ y S₂. Por principios de combinatoria, el número de secuencias diferentes que es posible generar a partir de S₁ (de longitud n) con r espacios es :
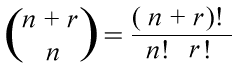
Dado que k! = 1×2×3×…k incluso para cadenas muy cortas, pongamos por ejemplo de 10 nucleótidos con 5 espacios, las variantes serían: (10+5)! ÷ 10! 5! = 3.003
Ahora hay que alinear la secuencia S₂ con S₁. Se puede comprobar que el número total de alineamientos posibles es:
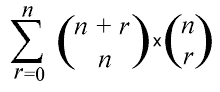
Es decir ¡el numero de alineamientos posibles entre dos secuencias crece vertiginosamente rápido a medida que aumenta la longuitud de las mismas!
Afortunadamente, no es necesario generar y puntuar todos los alineamientos para encontrar el óptimo. El truco consiste en componer una matriz con las secuencias en los ejes de abcisas y ordenadas. Con dicha disposición de las secuencias, es posible demostrar que cada alineamiento posible se corresponde con un camino entre la esquina superior izquierda ✲ y la inferior derecha ✲. Para trazar estos caminos se puede recorrer la diagonal si los nucleótidos de la abcisa y la ordenada coinciden y si no coinciden se debe hacer un desplazamiento vertical u horizontal, lo cual equivale a insertar un espacio en una u otra secuencia. Cuando se puede recorrer la diagonal se suma 1 al camino que se está recorriendo y cuando hay que moverse verticalmente u horizontalmente se resta 1. El alineamiento (o alineamientos) óptimo según la métrica que hemos descrito anteriormente es aquel que tiene la diagonal más larga. En el caso del camino azúl de la matriz mostrada su puntuación será -1+1-1+1-1+1+1+1+1+1+1+1=6.
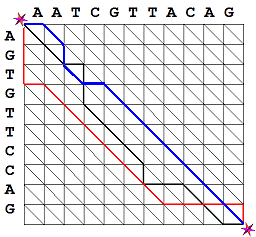
No voy a detallar aquí el algoritmo de programación dinámica basado en explorar el vecindario de cada nodo ┼ para trazar el camino y seguidamente reconstruir el alineamiento a partir del camino. Lo importante es que dicho algoritmo se puede ejecutar en un tiempo proporcional a 12×m×n, Siendo m y n la longuitud de las secuencias. Es decir, para dos secuencias de igual longuitud n la complejidad del algoritmo para alinearlas globalmente es O(n²) lo cual sigue siendo computacionalmente costoso pero es un tiempo polinómico en lugar de la horrenda expresión factorial.
El alineamiento global se usa para comparar genes. Incluso entre diferentes especies. Si identificamos un gen de ratón con una determinada función biológica (recordemos que un gen codifica una proteína) entonces podemos buscar un gen parecido en los humanos y si lo encontramos probablemente tendrá una función biológica en los humanos muy similar a la que tiene en los ratones.
Otro problema de alineamiento relacionado es que dadas dos secuencias S₁ y S₂ de longuitudes m y n, se desea encontrar dos subsecuencias de longitudes p y q tales que el valor de su alineamiento sea máximo. El algoritmo de Smith–Waterman para el alineamiento local es una variante del método de buscar la diagonal más larga excepto que permite empezar y terminar por nodos diferentes del superior izquierdo y el inferior derecho, y en un momento dado se puede descartar un conjunto de nodos ya explorados porque su puntuación sea negativa.
Respecto del ensamblado de secuencias cortas (entre 500 y 800 nucleótidos) para secuenciar genomas enteros con millones de pares de bases, buscar el mejor encaje entre el final de una secuencia y el principio de otra tomada de un conjunto de candidatas, es un problema muy similar a buscar un alineamiento global entre el final de una secuencia y el principio de otra excepto porque en este caso podemos esperar que el alineamiento, en el caso de existir, tenga muy pocas no coincidencias (debido a que las dos subsecuencias son trozos de la misma secuencia original) excepto porque se producen esporádicamente errores en la lectura de los nucleótidos. Con los secuenciadores modernos, la tasa de errores no supera el 0,1%, por consiguiente, el algoritmo deberá aplicar alguna técnica de corrección de errores y luego penalizar mucho las no-coincidencias.
Por último, puede presentarse el problema de encontrar el mejor alineamiento múltiple para una colección s de secuencias de longuitud n. La complejidad de este problema es O(ns) y, de hecho, se sabe que es un problema NP-Completo, por consiguiente, no existe ninguna solución que se pueda encontrar en tiempo polinómico. Sin embargo, el alineamiento de múltiples secuencias puede proporcionar información que es imposible de obtener con el alineamiento de sólo dos secuencias. Sucede que en regiones de ADN con funciones similares las partes de mayor importancia evolucionan con mayor lentitud que las partes menos relevantes biológicamente. Entonces es posible tomar, por ejemplo, secuencias de diferentes mamíferos, perros, gatos, ratones, humanos, etc. e identificar qué subsecuencias cambian (o no cambian) en operones que se sabe que cumplen funciones biológicas parecidas. Recíprocamente, si se tiene una secuencia que no se sabe para qué sirve pero puede alinearse de forma múltiple con otras secuencias de una colección que sí se sabe para qué sirven, entonces es posible concluir que la secuencia desconocida pertenece probablemente a la colección con la que tiene un buen alineamiento múltiple.
Una colección de secuencias relacionadas mediante un alineamiento múltiple se conoce como un perfil. Cuando se quiere saber si una nueva secuencia se ajusta a un determinado perfil, lo que se puede hacer es comparar el nucleótido en cada posición con los nucleótidos en la misma posición en el perfil, teniendo en cuenta que el perfil da una probabilidad para nucleótido A,T,C,G en la posición n. No obstante, existen otros métodos para determinar si una secuencia tiene un determinado perfil, en particular, los modelos de Markov que veremos a continuación.
Modelos de Markov
Resumidamente, un modelo de Markov consiste en un conjunto de estados y una probabilidad de transición de un estado a otro. Para el ADN tomaremos cuatro estados: A,T,C,G. A cada transición de un estado a otro A→A, A→T, A→C, A→G, T→A, T→T, etc. se le asigna una probabilidad. En este caso, si las transiciones de estado fuesen completamente aleatorias, la probabilidad de transición desde cada estado al siguiente sería ¼. Una secuencia de transiciones de estado contruida siguiendo las probabilidades de transición de un estado a otro se denomina una cadena de Markov. El generador puede representarse como un grafo dirigido con una probabilidad asociada a cada una de las aristas.
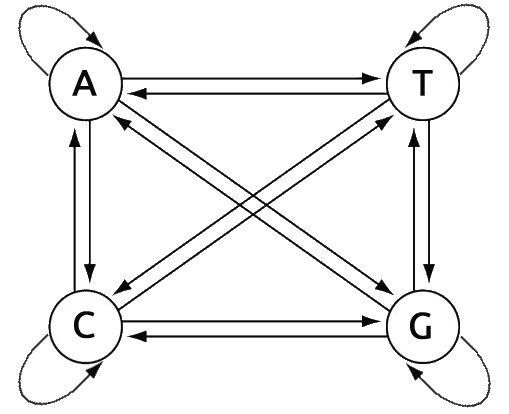
Esto tiene varias aplicaciones en genómica. Sucede, por ejemplo, que las regiones CpG (un nucleótido C seguido de otro G) son raras excepto para indicar el comienzo de un gen, con una longitud de la región CpG desde cien a unos pocos miles de pares de bases.
|
Probabilidades de transición en regiones CpG.
|
Probabilidades de transición en regiones no CpG.
|
||||||||||||||||||||||||||||||||||||||||||||||||||
| Fuente: Hidden Markov Model for CpG islands | |||||||||||||||||||||||||||||||||||||||||||||||||||
Con estos datos es posible comparar la probabilidad de que una secuencia sea una región CpG versus una región no CpG. Para ello basta con multiplicar las probabilidades de transición. Por ejemplo, dada la secuencia CGACGT en cada modelo habría que multiplicar: C→G × G→A × A→C × C→G × G→T. Entonces la probabilidad de que CGACGT haya sido generada por el modelo CpG es 0,274 × 0,161 × 0,274 × 0,274 × 0,125 = 0,000414 y para el modelo no CpG es 0,078 × 0,248 × 0,300 × 0,078 × 0,208 = 0,000094. Es decir, es cuatro veces más probable que la secuencia CGACGT pertenezca a una región CpG que a una región no CpG.
Llegados a este punto, es fácil intuir que dada una secuencia querremos hallar el Modelo de Markov Oculto que la generó. Es decir, la Cadena de Markov que con una mayor probabilidad generará la secuencia que ya tenemos. Esto se hace con el algoritmo de Viterbi, otro algoritmo de programación dinámica cuyos detalles no comentaré aquí.
En resumen, la clave del Modelo de Markov es que la probabilidad de encontrar cada estado se calcula exclusivamente en base al estado inmediatamente anterior. Aquí lo hemos ilustrado con nucleótidos, pero también puede usarse un modelo de Markov con codones en cuyo caso tendríamos sesenta y cuatro estados en lugar de cuatro y la probabilidad de encontrar un codón dependería del codón inmediatamente anterior.
Filogenética y evolución del genoma
La filogenética computacional es el área de la genómica computacional que se ocupa de estudiar la relación entre especies o taxones (grupos de organismos emparentados). Supongamos que tenemos cinco especies: A,B,D,E y F. El primer desafío es definir qué caracteriza a una especie. Esto se hace, en principio, mediante la observación externa. Y a continuación habrá que determinar qué variaciones en el ADN causan la biodiversidad. Esta labor de caracterización de las especies no es nada sencilla, pero, por el momento, vamos a suponer que, tras una árdua labor, ya la tenemos hecha para nuestras cinco especies. Además presumiremos la existencia de algún tipo de reloj molecular que marca el ritmo de la evolución. Con estas hipótesis una posible forma (muy simplificada) de modelar la evolución es mediante árboles ultramétricos. En un árbol ultramétrico, cada nodo interno tiene exactamente dos sucesores y un número estrictamente decreciente desde la raíz.
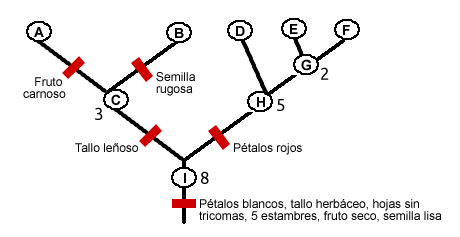
Este árbol expresa que las especies A y B surgieron a partir de la C (posiblemente extinta) hace 3 unidades de tiempo. Y la la D y la G surgieron de la H hace 5 unidades de tiempo. En la práctica, los números en los nodos internos del árbol se obtienen normalmente por alineamiento de secuencias, es decir, se asume que el grado de variación entre especies está correlacionado con el tiempo.
Para la búsqueda de un árbol filogenético que requiera el menor número de eventos evolutivos (reconstrucción filogenética) pueden emplearse diversos métodos: máxima parsimonia, máxima verosimilitud, inferencia bayesiana (IB) y matriz de distancias son los más comunes que por brevedad no describiremos aquí. El lector interesado puede encontrar más detalles sobre filogenética y modelos de Markov en el texto clásico Biological Sequence Analysis.
Búsquedas en bases de datos de secuencias
Debido a que el alineamiento de secuencias es computacionalmente costoso (proporcional al producto de la longitud de las secuencias), se necesita alguna técnica heurística para buscar una secuencia recientemente obtenida en una base de datos de secuencias conocidas. Especialmente si existe un elevado número de usuarios buscando concurrentemente alineamientos en una base de datos.
La primera herramienta que se popularizó para buscar secuencias relacionadas fue BLAST (Basic Local Alignment Search Tool). La idea de la primera versión de BLAST fué que si dos secuenciias son similares –en el sentido en el que hemos definido la similutud por la existencia de un alineamiento con una buena puntuación en la métrica anterior– entonces existirán subcadenas de nucleótidos idénticas entre las dos secuencias. Por ejemplo:
AACTCCAATAGGAA
La hipótesis de BLAST es que si en dos secuencias no es posible encontrar una cierta cantidad de subcadenas idénticas de suficiente longitud, entonces es poco probable que exista ningún buen alineamiento entre las secuencias. BLAST sólo proporciona resultados probabilísticos porque es fácil construir ejemplos de secuencias que no cumplen la condición de BLAST y que en cambio se pueden alinear bien insertando espacios y usando matrices de substitución. Por “suficiente longuitud” de las subcadenas idénticas se entiende aquella que hace poco probable que la coincidencia sea casual. Como hay 4 bases A,T,C,G entonces la probabilidad de que dos nucleótidos cualesquiera sean iguales por casualidad es del 25%. Por eso, para el ADN, se suelen tomar subcadenas de longuitud a partir de 11. O al menos así era en las primeras versiones de BLAST, porque comparando los candidatos que devolvía BLAST con los mejores alineamientos locales hallados mediante Smith-Waterman se comprobó que una opción mejor era acortar las subcadenas con la condición adicional del que se pudieran encontrar muchas de ellas próximas las unas a las otras. No voy a detallar aquí las razones estadísticas por las cuales BLAST selecciona secuencias candidatas a ser similares a una secuencia dada, dejémoslo en que para la secuencia buscada BLAST proporciona una lista de resultados ordenados por puntuación P y, además de P, BLAST proporciona otro valor llamado E que indica la probabilidad de encontrar la misma o mayor cantidad de resultados con puntuación P para la secuencia buscada si la base de datos estuviese compuesta de secuencias puramente aleatorias.
Bases de datos de ADN y proteinas
Cuando se secuencia una nueva cadena de ADN, se compara mediante alineamiento con otras secuencias conocidas y se añaden anotaciones que señalan la localización de los genes y el resto de regiones que controlan lo que hacen los genes: dónde empiezan y terminan intrones y exones, secuencias reguladoras, repeticiones, función biológica, expresión etc. Existen herramientas para anotar automáticamente que suelen usarse en combinación con anotaciones realizadas manualmente.
El resultado de la secuenciación, alineamiento y anotación se almacena en una base de datos. Existen bastantes bases de datos públicas y privadas. Sólo en Wikipedia puede encontrarse una lista con 180 bases de datos de ADN y proteínas. La más popular es la International Nucleotide Sequence Database (INSD) que agrupa la japonesa DNA Data Bank of Japan, la norteamericana GenBank y la europea EMBL-EBI. Existen también bases de datos específicas de genes que causan enfermedades y bases de datos de expresión genética.
Software disponible
Hitóricamente, el lenguaje de referencia para aplicaciones de bioinformática era Perl, aunque con el paso de los años han ido apareciendo herramientas en otros lenguajes como Java, R y Python. EMBOSS, posiblemente la herramienta más popular para realizar alineamiento de secuencias está escrito principalmente en C. Incluso hay una distro de Ubuntu llamada Bio-Linux que contiene más de 250 programas libres para bioinformática.
Lo típico es usar una base de datos de secuencias basada en BLAST como la del EMBL-EBI, un analizador-ensambldor como EMBOSS o Staden, un programa de predicción de estructura protéica como THREADER o un programa de modelado molecular estilo RasMol.
Incluso Google ofrece su plataforma cloud con fines específicos de genómica computacional.
En general, la caja de herramientas del bioinformático contendrá más o menos lo siguiente:
• Análisis de secuencias, mutaciones, regiones, etc.
• Cálculo de similitud y homología de secuencias, ancestros, etc.
• Análisis estructural 2D/3D de proteínas.
• Análisis de función de proteínas (a partir de su estructura).
En Wikipedia se puede encontrar una lista de software Open Source para bioinformática. También existen listados de proveedores de software y servicios de genómica computacional y listados de las principales empesas de bioinformática.
Hardware de propósito específico
El tratamiento informatizado de la información genética es un desafío complejo que tiende a alcanzar los límites del hardware actual, tanto en uso de CPU como en consumo de memoria y capacidad de almacenamiento. Prácticamente desde que se inventaron las FPGA (Field Programmable Gate Array) hace treinta años, se sabe que pueden usarse en lugar de CPUs para acelerar la ejecución de algoritmos de alineamiento de secuencias. La clave es que el algoritmo de Smith-Waterman se puede paralelizar parcialmente y, además, sólo requiere aritmética de 24 bits. Entender cómo se consigue acelerar la ejecución mediante el paralelismo requeriría entrar en los detalles del algoritmo, cosa que no hemos hecho, por consiguiente el lector interesado puede referirse a artículos como éste.
La empresaa pionera en hardware específico para el alineamiento de secuencias es Timelogic, fundada en 1981 y adquirida en 2003 por Active Motif. Luego se sumaron otras como SGI y, porsupuesto, los fabricantes de hardware de toda la vida con las mismas máquinas pero con una etiqueta nueva que pone «cluster de bioinformática».
Retos actuales
Es dificil enumerar los retos en un área como la genómica computacional donde todavía está prácticamente todo por hacer. Actualmente es posible secuenciar ADN de forma económicamente viable pero en el ensamblado de las lecturas cortas producidas por los secuenciadores como Illumina todavía existen problemas abiertos relacionados con las repeticiones y la fase.
Analizar el amplísimo rango de fenotipos con la limitada base de la información disponible sobre el genoma humano es otro desafío.
El entendimiento de cómo funcionan las secciones reguladoras es aún escasísimo.
Y existen innumerables problemas técnicos que no tienen que ver con el tratamiento informatizado sino con el hecho de que el ADN funciona a nivel molecular y, por consiguiente, es demasiado pequeño cómo para poderlo manipular directamente.
Todo ello no menoscaba el potencial de la bioinformática, pues simplemente estamos en los albores de un salto cuántico hacia adelante en nuestro entendimiento sobre cómo funciona la vida.